


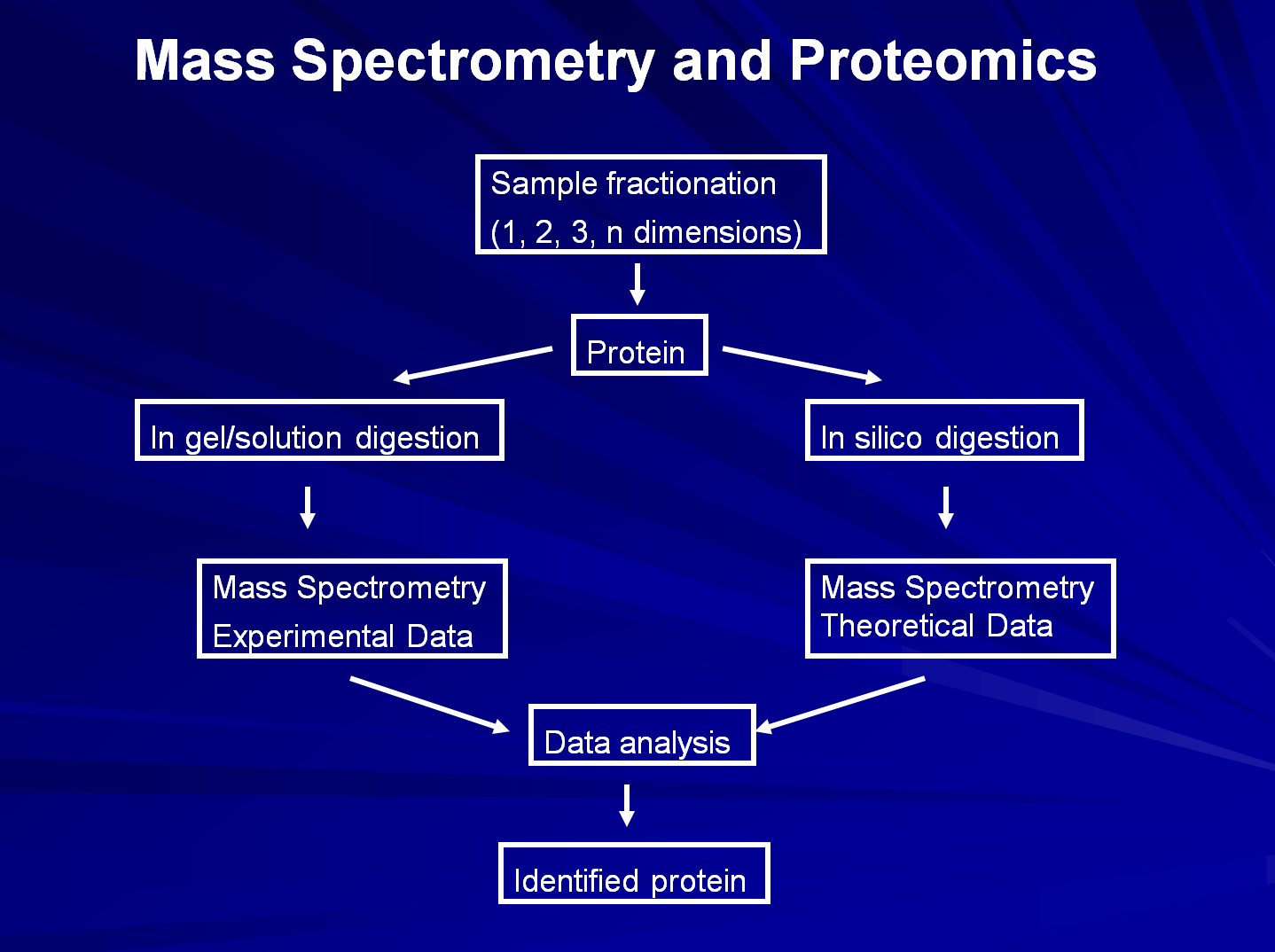
The workflow in a proteomics experiment involves sample fractionation by 1, 2, n biochemical approaches, followed by enzymatic digestion (usually trypsin), peptide extraction, and MS analysis (1). When the peptide mixture is analyzed by MALDI-MS, the proteins of interest are identified using a procedure named peptide mass fingerprinting. Alternatively, the peptide mixture is further fractionated by HPLC on different columns (usually reverse phase HPLC or LC), followed by ESI-MS analysis. The combination of LC and ESI-MS is usually named LC-MS/MS, and analysis of a protein using this approach provides not only the protein identity, but also sequence information for that particular protein.




HTML and CSS written by Christopher Talbot, Clarkson University 2010